#1 Excellent
100AGACCGTGGACAAGATCGAG
PAM TGG, strand +, coordinates 5–24, GC 55.0%.
GC% sits in the preferred 40–60% range.
Find guide RNA candidates from a DNA sequence, locate PAM sites, and rank sgRNAs with transparent first-pass rules. The tool helps students and researchers shortlist guides before genome-wide off-target analysis.
Start with SpCas9 NGG in Basic mode. Switch to Advanced mode for Cas12a, SaCas9, SpG, custom IUPAC PAMs, and cloning filters.
Basic mode finds common PAMs. Advanced mode exposes custom IUPAC patterns and guide filters.
Paste genomic DNA or a PCR amplicon. The scanner removes spaces and ignores non-DNA characters.
Clean length
153
bp scanned
Select the nuclease, PAM pattern, and cloning filters that match your experimental plan.
Live guide screen
Best candidate: AGACCGTGGACAAGATCGAG next to PAM TGG. Score 100/100 with 55.0% GC.
Total hits
18
Excellent
18
Good
0
Review
0
The map shows the strongest candidates across your submitted sequence. Purple marks the PAM-side end.
Review top guides first, then confirm specificity with a genome-aware off-target search.
AGACCGTGGACAAGATCGAG
PAM TGG, strand +, coordinates 5–24, GC 55.0%.
GC% sits in the preferred 40–60% range.
GTGCCACTCGATCTTGTCCA
PAM CGG, strand -, coordinates 11–30, GC 55.0%.
GC% sits in the preferred 40–60% range.
GGACAAGATCGAGTGGCACG
PAM AGG, strand +, coordinates 12–31, GC 60.0%.
GC% sits in the preferred 40–60% range.
GTGGCACGAGGACCTGTTCA
PAM AGG, strand +, coordinates 24–43, GC 60.0%.
GC% sits in the preferred 40–60% range.
GGACCTGTTCAAGGCCATCG
PAM TGG, strand +, coordinates 33–52, GC 60.0%.
GC% sits in the preferred 40–60% range.
GCTCCACGATGGCCTTGAAC
PAM AGG, strand -, coordinates 39–58, GC 60.0%.
GC% sits in the preferred 40–60% range.
CCATCGTGGAGCAGTACGAG
PAM CGG, strand +, coordinates 47–66, GC 60.0%.
GC% sits in the preferred 40–60% range.
CGGATCGTGAAGCTGCTGAC
PAM CGG, strand +, coordinates 67–86, GC 60.0%.
GC% sits in the preferred 40–60% range.
ATCGTGAAGCTGCTGACCGG
PAM CGG, strand +, coordinates 70–89, GC 60.0%.
GC% sits in the preferred 40–60% range.
GAACTCGTCGATGTAGCCGC
PAM CGG, strand -, coordinates 89–108, GC 60.0%.
GC% sits in the preferred 40–60% range.
CTACATCGACGAGTTCATCG
PAM AGG, strand +, coordinates 93–112, GC 50.0%.
GC% sits in the preferred 40–60% range.
CGAGTTCATCGAGGACGCCA
PAM AGG, strand +, coordinates 102–121, GC 60.0%.
GC% sits in the preferred 40–60% range.
AGGACGCCAAGGAGATCGTG
PAM CGG, strand +, coordinates 113–132, GC 60.0%.
GC% sits in the preferred 40–60% range.
CAAGGAGATCGTGCGGCTGA
PAM AGG, strand +, coordinates 120–139, GC 60.0%.
GC% sits in the preferred 40–60% range.
CTTCAGCCGCACGATCTCCT
PAM TGG, strand -, coordinates 122–141, GC 60.0%.
GC% sits in the preferred 40–60% range.
CCGCTCGTACTGCTCCACGA
PAM TGG, strand -, coordinates 50–69, GC 65.0%.
GC% is acceptable but outside the strongest 40–60% band.
GGAGATCGTGCGGCTGAAGG
PAM AGG, strand +, coordinates 123–142, GC 65.0%.
GC% is acceptable but outside the strongest 40–60% band.
GATCGTGCGGCTGAAGGAGG
PAM AGG, strand +, coordinates 126–145, GC 65.0%.
GC% is acceptable but outside the strongest 40–60% band.
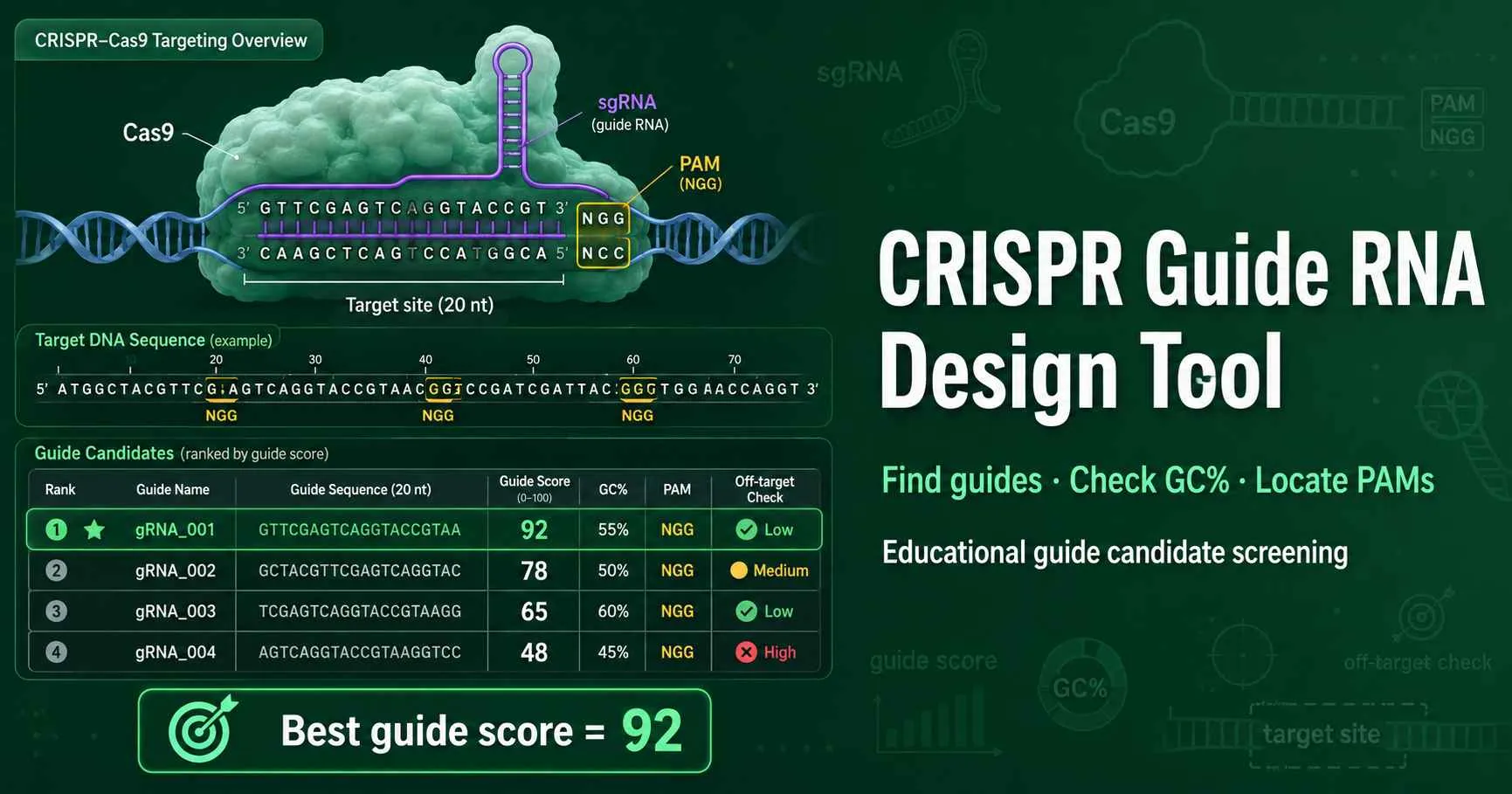
Use this page when you have a target DNA region and need candidate guide RNAs quickly. Paste the sequence, choose a nuclease, and review guides beside valid PAM sites. For a PAM-only scan without guide ranking, use the PAM Sequence Finder first.
The calculator scores sequence features that researchers check before ordering guides: GC percentage, homopolymer runs, U6-compatible 5′ G, and poly-T motifs. It also maps candidates on both DNA strands and reports original sequence coordinates.
Addgene explains that CRISPR specificity depends on both the guide RNA sequence and the Cas enzyme, and that partial homology elsewhere can create off-target activity. The 2016 Doench sgRNA design paper built empirical rules to improve on-target activity and reduce off-target effects in genome-scale libraries. Read Addgene’s CRISPR guide.
Each field supports one decision in a real guide-selection workflow. Basic users can keep defaults. Advanced users can narrow a chosen exon or test a non-SpCas9 PAM.
Provides the source region for protospacer and PAM scanning. Paste the coding strand, genomic strand, or amplicon sequence.
Defines PAM pattern, guide length, PAM side, and approximate cut-site position for SpCas9, Cas12a, SaCas9, SpG, or custom enzymes.
Restricts guide candidates to a chosen exon, domain, enhancer, promoter segment, or classroom example region.
Flags guides that may need an added G for U6 promoter expression or cloning into a specific vector.
Warns when a TTTT motif may stop Pol III transcription before the full sgRNA sequence forms.
Shows guide DNA, guide RNA, PAM, strand, coordinates, GC%, score, and warnings for export or review.
The tool rewards guides near 40–60% GC. That range often balances target binding with manageable synthesis and secondary-structure risk. If your target region has extreme base composition, compare guide candidates with the GC Content Calculator before you choose a final set.
SpCas9 recognition depends strongly on the PAM and nearby seed region. This page flags extreme seed GC content and reports the PAM-side end on the map. Hsu and colleagues showed that mismatch effects depend on mismatch number, position, and distribution. View the Cas9 specificity study.
| Check | Preferred value | Why it matters |
|---|---|---|
| Guide length | 20 nt for SpCas9 | Matches common SpCas9 protospacer design. |
| PAM | NGG for SpCas9 | Cas9 must recognize a nearby PAM before stable target interrogation. |
| GC% | 40–60% | Balances weak AT-rich guides against overly stable GC-rich guides. |
| Poly-T | Avoid TTTT | Pol III promoters can terminate at T-rich runs. |
A student pastes a 180 bp coding exon and selects SpCas9 NGG. The tool finds several 20 nt guides on both strands. A guide with 52% GC, no TTTT motif, and no 4-base homopolymer earns a strong first-pass score.
The student still checks transcript isoforms, exon usage, and genome-wide specificity before ordering. They also design PCR primers around the cut site and test those primers with the Primer Dimer Calculator.
A researcher wants guides in an AT-rich promoter. SpCas9 returns few NGG sites, so they switch to Cas12a TTTV. The tool now scans PAMs upstream of 23 nt guide regions and reports both-strand coordinates.
Cas12a candidates may suit AT-rich targets, but the final choice still depends on the nuclease, delivery system, organism, and genome-wide off-target profile.
This page gives a transparent first-pass screen. It does not search a whole genome. Off-target risk depends on near matches outside your pasted sequence, mismatch position, chromatin context, guide concentration, nuclease variant, and delivery method.
Use at least two independent guides when possible. Validate editing at the DNA level, confirm transcript or protein disruption when needed, and include non-targeting or safe-targeting controls. Never use this educational tool as clinical guidance.
Use these tools before or after guide selection to check PAM positions, sequence properties, and oligo behavior.
Scan DNA for NGG, NAG, TTTV, and custom IUPAC PAM patterns before guide ranking.
Check sequence length, GC content, reverse complement, ORFs, and restriction sites.
Review short guide-adaptor oligos or validation primers for complementarity risk.
It scans a DNA sequence for PAM sites, extracts nearby guide sequences, and ranks candidate sgRNAs with simple sequence-quality rules. It reports guide coordinates, PAM sequence, strand, GC percentage, homopolymer warnings, poly-T warnings, and a first-pass score. The tool supports SpCas9 NGG, SpCas9 NAG, Cas12a TTTV, SpG NGN, SaCas9 NNGRRT, and custom IUPAC PAM patterns. It does not replace a genome-wide off-target search.
SpCas9 guide design usually uses a 20 nucleotide protospacer next to a 3′ NGG PAM. The tool extracts 20 bases upstream of each NGG site on the plus strand and performs the reverse-complement equivalent on the minus strand. It reports the guide DNA sequence and the RNA version with U in place of T. Use the Advanced mode only when your nuclease, vector, or classroom exercise uses a different guide length.
A practical first-pass guide often sits near 40–60% GC content. Very low GC can weaken target binding, while very high GC can increase secondary structure or synthesis difficulty. The score in this tool rewards guides inside that 40–60% window and flags guides far outside it. GC content alone never proves that a guide will cut well.
Many sgRNA expression cassettes use a U6 promoter. A run of four or more thymidines in the DNA guide can behave like a Pol III termination signal after transcription. The tool flags TTTT because it can reduce full-length sgRNA expression. This warning matters most for U6-driven guide expression and may not apply to every delivery system.
No. This page screens the sequence you paste, but it does not search a whole genome. True off-target analysis needs a reference genome, mismatch rules, genomic coordinates, and sometimes chromatin or cell-type context. Use this tool to shortlist candidates, then run genome-aware off-target checks before ordering guides. Experimental validation still matters.
Knockout experiments often target early constitutive coding exons because frameshift indels can disrupt more of the protein. The best target region depends on transcript isoforms, protein domains, exon usage, and whether the gene tolerates alternative start sites. The tool lets you restrict the scan to a target window, such as a chosen exon or domain. Confirm exon coordinates in a genome browser before final design.
The score gives a transparent first-pass ranking from sequence features. It rewards balanced GC content and penalizes homopolymers, TTTT motifs, missing 5′ G when requested, and extreme seed composition. It does not use a trained genome-wide model. Treat it as a classroom and planning score, not a clinical or regulatory guide-design decision.