How to Calculate Allele Frequency From Genotypes

To calculate an allele frequency from genotype counts, you count how many copies of that allele exist in the population and divide by the total number of alleles. For a gene with two alleles in a sample of N individuals, the dominant allele frequency is p = (2 × number of AA + number of Aa) / 2N, and the recessive allele frequency is q = (2 × number of aa + number of Aa) / 2N. Each individual carries two alleles, so the total is 2N. This direct counting method works for any population, whether or not it is in Hardy-Weinberg equilibrium.
Allele frequency is the starting point for all of population genetics, and counting it from genotype data is the most fundamental skill in the topic. This guide shows you both standard methods, the allele-counting method and the genotype-frequency method, with clear worked examples for each, so you can handle whatever form your data arrives in. It also explains a subtle but important point: you can always find allele frequencies from genotype counts directly, but the reverse requires an assumption. These calculations can be run instantly with a calculator, though knowing the method by hand makes the results meaningful.
What an Allele Frequency Is
Before calculating anything, it helps to be clear on what an allele frequency actually measures. An allele frequency is the proportion of all the copies of a gene in a population that are a particular allele. It answers the question: out of every copy of this gene in the whole population, what fraction is this version?
The key to understanding this is the gene pool concept. Imagine collecting every copy of a particular gene from every individual in a population into one large pool. Because most organisms are diploid, carrying two copies of each gene, a population of N individuals contributes 2N copies to the pool for that gene. The allele frequency is simply the fraction of that pool made up by each allele. If the pool holds 200 alleles and 150 of them are the A allele, then the frequency of A is 150 divided by 200, which is 0.75.
This framing makes the whole calculation intuitive. You are not measuring individuals; you are measuring alleles. An individual with the genotype AA contributes two A alleles to the pool, an individual with aa contributes two a alleles, and a heterozygous Aa individual contributes one of each. Once you see that every calculation is just counting alleles in this pool and dividing by the total, the formulas stop looking like something to memorize and become obvious. The frequencies of the two alleles must add up to 1, because together they make up the entire pool, which is the basis of the p + q = 1 relationship at the heart of population genetics.
Method 1: The Allele-Counting Method
The most reliable way to find allele frequencies is to count the alleles directly, and it works for any set of genotype data. This method does not assume the population is in equilibrium or anything else; it simply tallies the alleles present and divides by the total. That makes it the safest default.
The logic follows from how each genotype contributes to the gene pool. A homozygous individual contributes two copies of one allele, while a heterozygous individual contributes one copy of each. So to count all the A alleles, you take twice the number of AA individuals (each gives two A's) plus the number of Aa individuals (each gives one A). To get the frequency, you divide by the total number of alleles, which is 2N where N is the number of individuals. This gives the formula p = (2 × AA + Aa) / 2N for the dominant allele.
The recessive allele works the same way. You count twice the number of aa individuals plus the number of Aa individuals, then divide by 2N, giving q = (2 × aa + Aa) / 2N. As a built-in check, p and q must add up to 1, since every allele in the pool is either A or a. If your calculated p and q do not sum to 1, you have made a counting error somewhere. This counting method is the foundation, and it is worth becoming completely comfortable with it before moving to any shortcut.
A Worked Example of Counting Alleles
Numbers make the method concrete. Suppose you sample a population and find 320 individuals with genotype AA, 160 with genotype Aa, and 20 with genotype aa. The goal is to find the frequencies of the A and a alleles.
First, find the total number of alleles. The number of individuals is 320 plus 160 plus 20, which is 500. Since each individual has two alleles, the total number of alleles is 2 times 500, which is 1000. Now count the A alleles. The AA individuals contribute 2 times 320, which is 640 A alleles, and the Aa individuals contribute 160 A alleles, for a total of 800. So p, the frequency of A, is 800 divided by 1000, which is 0.8.

Now do the same for the a allele. The aa individuals contribute 2 times 20, which is 40 a alleles, and the Aa individuals contribute 160 a alleles, for a total of 200. So q, the frequency of a, is 200 divided by 1000, which is 0.2. Check the result: p plus q equals 0.8 plus 0.2, which is 1, exactly as it must. From a simple count of genotypes, you have found that 80 percent of the alleles in this population are A and 20 percent are a. This counting approach is direct, reliable, and the method to reach for whenever you have genotype counts.
Method 2: The Genotype-Frequency Method
A second method starts from genotype frequencies rather than raw counts, and it is useful when your data are already given as proportions or percentages. It reaches the same answer through a slightly different route, and understanding it deepens your grasp of where the numbers come from.
The principle is that an allele's frequency equals the frequency of its homozygote plus half the frequency of the heterozygote. In symbols, p equals the frequency of AA plus half the frequency of Aa, and q equals the frequency of aa plus half the frequency of Aa. The reason for the half is that heterozygotes carry only one copy of each allele, so they contribute only half their number to each allele's count. Homozygotes carry two copies of their allele, so they contribute their full frequency.
This method is just the counting method expressed in terms of proportions. If 64 percent of a population is AA, 32 percent is Aa, and 4 percent is aa, then p equals 0.64 plus half of 0.32, which is 0.64 plus 0.16, giving 0.80.
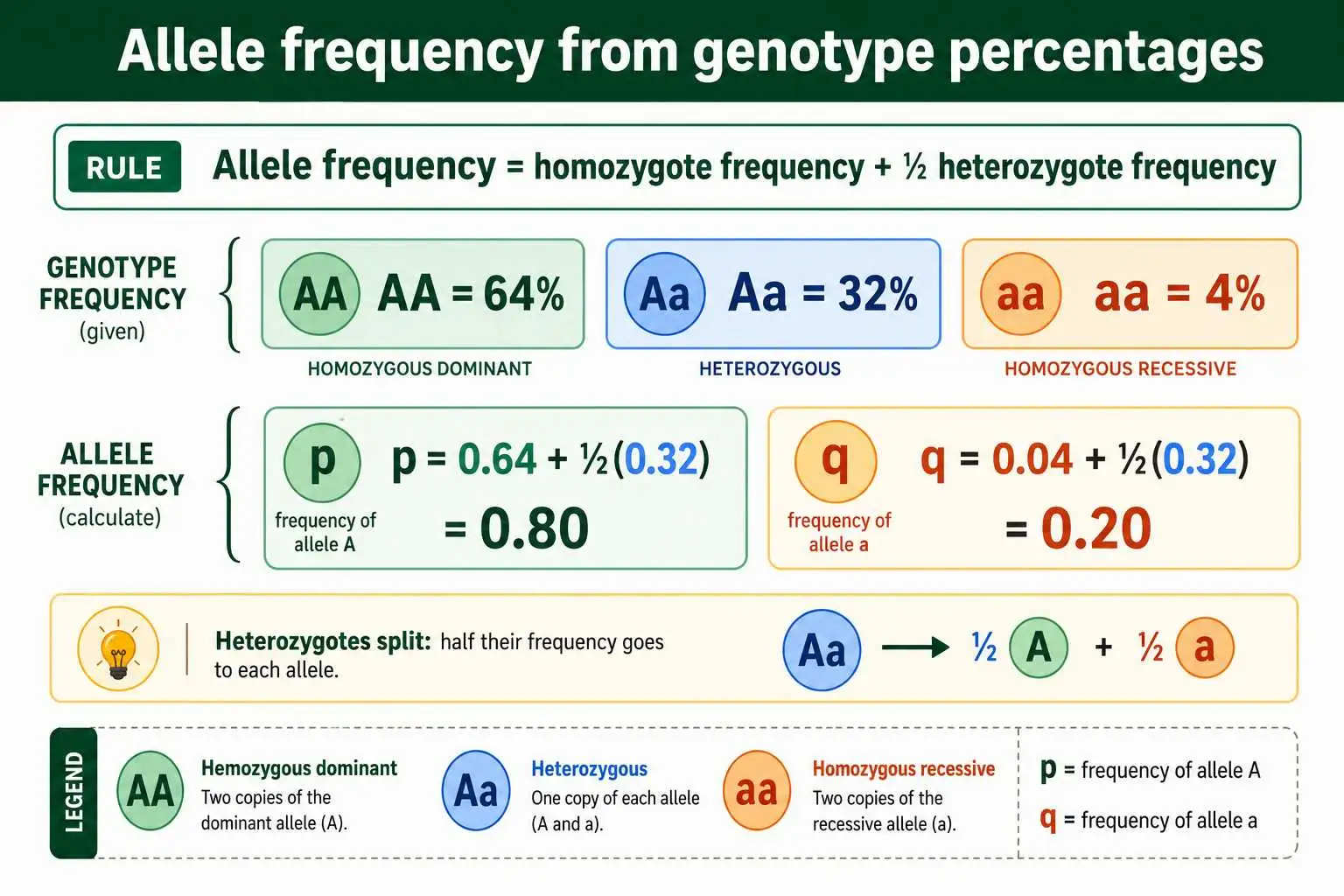
Likewise q equals 0.04 plus half of 0.32, which is 0.04 plus 0.16, giving 0.20. Again p and q sum to 1. Use this method when your data come as frequencies or percentages, and use the counting method when you have raw individual counts; they are two views of the same underlying idea.
The Crucial Point: Counting Always Works
There is an important and often-overlooked distinction that separates a real understanding of this topic from a superficial one. You can always calculate allele frequencies from genotype counts directly, no assumptions required, but you cannot always go the other way. Going from allele frequencies back to genotype frequencies requires the Hardy-Weinberg assumption of random mating.
Here is why this matters. The counting method works on whatever genotypes are actually present, in any population, evolving or not. If you can observe and count the genotypes, you can find the allele frequencies, full stop. This is a pure measurement, not a prediction. It does not matter whether the population mates randomly, experiences selection, or anything else. The alleles are there to be counted, and counting them is always valid.
The reverse is not true. If you know only the allele frequencies and want to predict the genotype frequencies, you must assume the population is in Hardy-Weinberg equilibrium, because the prediction uses the p², 2pq, q² formula that only holds under those conditions. Two populations with identical allele frequencies can have completely different genotype frequencies if one is not in equilibrium. This is why allele frequency is information you can always extract from genotypes, while genotype frequency is something you can only predict from alleles under specific assumptions. Keeping this direction clear, genotypes to alleles is always safe, alleles to genotypes needs the assumption, prevents a common conceptual mistake. The predictive direction is exactly what our guide on using the Hardy-Weinberg equation covers.
Calculating From Phenotype Data
Sometimes you cannot observe genotypes directly, only phenotypes, and this is where the methods connect to the Hardy-Weinberg equation. With complete dominance, the homozygous dominant and heterozygous individuals look identical, so you cannot simply count their alleles. A different approach is needed.
The trick is to start from the recessive phenotype, which corresponds to a single genotype. Because only homozygous recessive individuals show the recessive phenotype, their frequency gives you q² directly. From there you take the square root to find q, then use p + q = 1 to find p. This is the only way to estimate allele frequencies from phenotype data, and crucially, it requires assuming Hardy-Weinberg equilibrium, since you are using the q² relationship to work backward.
This is the key difference from the counting method. When you can see genotypes, you count alleles directly with no assumptions. When you can only see phenotypes, you must lean on the Hardy-Weinberg equation and its assumptions to estimate the frequencies. For example, if 16 percent of a population shows a recessive trait, then q² is 0.16, so q is the square root of 0.16, which is 0.4, and p is 0.6. Notice that this estimate is only as good as the assumption that the population is in equilibrium, which is a real limitation. The detailed procedure for these phenotype-based calculations, including finding carrier frequencies, is covered in our guide on calculating carrier frequency.
Multiple Alleles and a Quick Generalization
The counting method extends naturally to genes with more than two alleles, which is worth knowing since many real genes are not limited to two. The principle stays exactly the same: count the copies of each allele and divide by the total.
For a gene with three or more alleles, the frequency of any one allele is the frequency of its homozygote plus half the sum of the frequencies of all the heterozygotes in which it appears. The ABO blood group is the classic example, with three alleles in the population. To find the frequency of one allele, you count every copy of it, whether in a homozygote contributing two copies or in any heterozygote contributing one, and divide by the total number of alleles. The math is identical to the two-allele case, just with more genotype categories to tally.
The reassuring point is that no new concept is needed. Whether a gene has two alleles or ten, an allele frequency is always the count of that allele divided by the total allele count. The two-allele formulas are simply the most common case. Once you understand that you are counting alleles in a gene pool, you can handle any number of alleles by extending the same tally. This generalization is part of why the counting method is so reliable: it rests on nothing more than counting, which always works regardless of how many alleles or what forces act on the population.
To make the multi-allele case concrete, consider a simplified ABO example. Suppose a sample contains individuals who are genotype IᴬIᴬ, IᴬIᴮ, IᴮIᴮ, Iᴬi, Iᴮi, and ii. To find the frequency of the Iᴬ allele, you count two copies for every IᴬIᴬ individual and one copy for every IᴬIᴮ and every Iᴬi individual, then divide by the total number of alleles in the sample. The same tally finds Iᴮ and i, and all three frequencies sum to 1. As the Wikipedia entry on allele frequency notes, the frequency of each allele is its homozygote frequency plus half the sum of every heterozygote in which it appears, which is exactly the two-allele rule extended to more alleles.
Common Mistakes When Calculating Allele Frequencies
A few predictable errors trip people up when finding allele frequencies, and all of them are easy to avoid once you know to watch for them. Catching these is the difference between a reliable answer and a confident wrong one.
The most common mistake is forgetting that each individual contributes two alleles, and dividing by N instead of 2N. The denominator must be the total number of alleles, not the total number of individuals, so a sample of 500 people has 1000 alleles for the gene. A second frequent error is mishandling heterozygotes, either counting them twice for one allele or forgetting they contribute to both allele totals. Each heterozygote adds exactly one copy of each allele, so it appears once in the count for A and once in the count for a. Treating it as contributing two of the same allele, the way a homozygote does, throws off the result.
A third mistake is confusing allele frequency with genotype frequency. The frequency of the a allele is not the same as the frequency of the aa genotype; the allele frequency counts every a, including those hidden in heterozygotes, while the genotype frequency counts only the aa individuals. Mixing these up is a conceptual error that leads to wrong answers throughout population genetics. The simplest safeguard against all three mistakes is the sum check: calculate both p and q independently and confirm they add to 1. If they do not, you have miscounted somewhere, and the error is usually one of these three.

Why Sample Size and Representativeness Matter
Calculating an allele frequency is only as good as the sample you calculate it from, a point that is easy to overlook when the arithmetic itself is simple. The formulas give a precise number, but that number describes your sample, and it represents the whole population only if the sample is large and unbiased.
Sample size matters because allele frequencies estimated from a small sample are unreliable. If you count genotypes in just ten individuals, a single unusual individual can swing the frequency dramatically, and your estimate may be far from the true population value. Larger samples average out this random variation and give estimates closer to reality. This connects to a deeper idea in population genetics: small populations are themselves subject to large random swings in allele frequency, an effect called genetic drift, which is one of the forces that pushes populations away from equilibrium.
Representativeness matters just as much as size. If your sample is biased, for instance, if you sampled only one family, one region, or one age group, the allele frequencies you calculate will reflect that subgroup rather than the whole population. A representative sample is drawn so that every part of the population has a fair chance of being included. For the counting method to give a meaningful population frequency, the sample must be both large enough to be stable and representative enough to be unbiased. The arithmetic cannot fix a poor sample, so good data collection is as important as correct calculation.
Frequently Asked Questions
What is the formula for allele frequency from genotypes?
For two alleles, the dominant allele frequency is p = (2 × AA + Aa) / 2N and the recessive is q = (2 × aa + Aa) / 2N, where AA, Aa, and aa are the counts of each genotype and N is the number of individuals. You count allele copies and divide by the total, 2N.
Why is the denominator 2N?
Because each individual is diploid and carries two alleles for the gene. A population of N individuals therefore has 2N total alleles for that gene. Dividing by 2N converts the allele count into a frequency, a proportion of the whole gene pool.
Do I need Hardy-Weinberg equilibrium to calculate allele frequency?
No, not when you have genotype counts. The counting method works for any population by simply tallying alleles. You only need the Hardy-Weinberg assumption when estimating allele frequencies from phenotype data, where you start from q² and work backward.
How do you find allele frequency from genotype percentages?
Use the genotype-frequency method: an allele's frequency equals its homozygote frequency plus half the heterozygote frequency. So p equals frequency of AA plus half the frequency of Aa, and q equals frequency of aa plus half the frequency of Aa.
Count First, Then Predict
Calculating allele frequency from genotypes comes down to counting. Add up the copies of an allele, with homozygotes contributing two and heterozygotes one, then divide by the total of 2N alleles. The formulas p = (2 × AA + Aa) / 2N and q = (2 × aa + Aa) / 2N capture this directly, and the genotype-frequency method gives the same answer from proportions. Always check that p and q sum to 1, since that single test catches most counting errors before they spread into later calculations.
The most important idea to carry forward is that counting alleles from genotypes always works, with no assumptions, while predicting genotypes from alleles, or estimating alleles from phenotypes, requires Hardy-Weinberg equilibrium. Master the counting method first, since it is the bedrock of everything else in population genetics. Every more advanced calculation, from predicting genotype frequencies to testing for equilibrium to measuring evolutionary change, begins with an accurate allele frequency, so the few minutes spent getting comfortable with counting pay off across the entire subject. You can verify any calculation with the Hardy-Weinberg allele frequency calculator, which counts the alleles and computes p and q for you. For a clear academic walkthrough of these methods, this population genetics chapter from Biology LibreTexts is a reliable reference.