What Affects the Mutation Rate?

The mutation rate is not a fixed constant. It is shaped by how faithfully DNA is copied, how well damage is repaired, how much mutagenic exposure a cell faces, the local DNA sequence, the age and sex of the parent, and, over evolutionary time, the size of the population. Each of these pushes the rate up or down, which is why mutation rates vary across genomes, individuals, and species.
This guide walks through the main factors. It covers replication fidelity and repair, mutagens, sequence hotspots like CpG sites, the paternal age effect, and the drift-barrier hypothesis that explains why the rate sits where it does. For the quantity itself, our explainer on what mutation rate is sets it up.
Replication Fidelity
The first thing that sets the mutation rate is how accurately DNA is copied during replication. Most mutations are copying errors, so the accuracy of the copying machinery is the primary control.
DNA polymerases, the enzymes that copy DNA, are remarkably accurate on their own, but their key feature is proofreading. Many polymerases have a built-in proofreading function, an exonuclease activity that checks each newly added base and removes it if it is wrong before moving on. Proofreading improves accuracy by roughly a hundredfold over the polymerase's raw error rate. Organisms or variants with weaker proofreading have higher mutation rates, and engineered proofreading-deficient polymerases are dramatically more error-prone.
So replication fidelity is the baseline. A cell that copies its DNA more accurately, through better polymerases and stronger proofreading, has a lower mutation rate. This is why mutations in the proofreading machinery itself can raise the genome-wide rate, producing what are called mutator phenotypes.
DNA Repair
After replication, DNA repair systems catch and fix errors and damage that slipped through. Repair is the second major control on the mutation rate, and it is extensive.
Hundreds of genes are devoted to finding and correcting DNA errors. Mismatch repair scans newly replicated DNA for mispaired bases and corrects them, adding another large boost to fidelity beyond proofreading. Other pathways fix specific kinds of damage: base excision repair handles small chemical lesions, nucleotide excision repair removes bulky distortions like those from UV light, and double-strand break repair stitches broken chromosomes back together. Each pathway that fails or weakens raises the mutation rate.
The importance of repair shows up starkly in disease. People with inherited defects in repair genes have elevated mutation rates and correspondingly high cancer risk, because unrepaired mutations accumulate in their cells. The condition xeroderma pigmentosum, a defect in repairing UV damage, leaves patients extremely vulnerable to skin cancer, a direct demonstration that repair capacity sets the somatic mutation rate. You can see how a change in the per-base rate scales up across a genome by comparing per-genome totals at different fidelity levels, which makes the effect of better or worse repair concrete.
Mutagens
External agents that damage DNA, called mutagens, raise the mutation rate. They are the most familiar factor because exposure is something individuals can sometimes control.
Mutagens come in several forms. Ultraviolet light from the sun creates bonds between adjacent DNA bases, causing the mutations behind most skin cancers. Ionizing radiation, from X-rays or radioactive sources, breaks DNA strands directly. Chemical mutagens, including many found in tobacco smoke, certain industrial compounds, and some natural toxins, react with DNA bases and cause mispairing. Even normal metabolism produces reactive oxygen species that damage DNA from within.
The common thread is that mutagens increase the damage load faster than repair can keep up, so more errors become permanent mutations. The effect is dose-dependent: more exposure means more mutations. This is why mutagen exposure matters for cancer risk, since somatic mutations drive tumor formation, and why limiting exposure to known mutagens is a basic cancer-prevention measure.
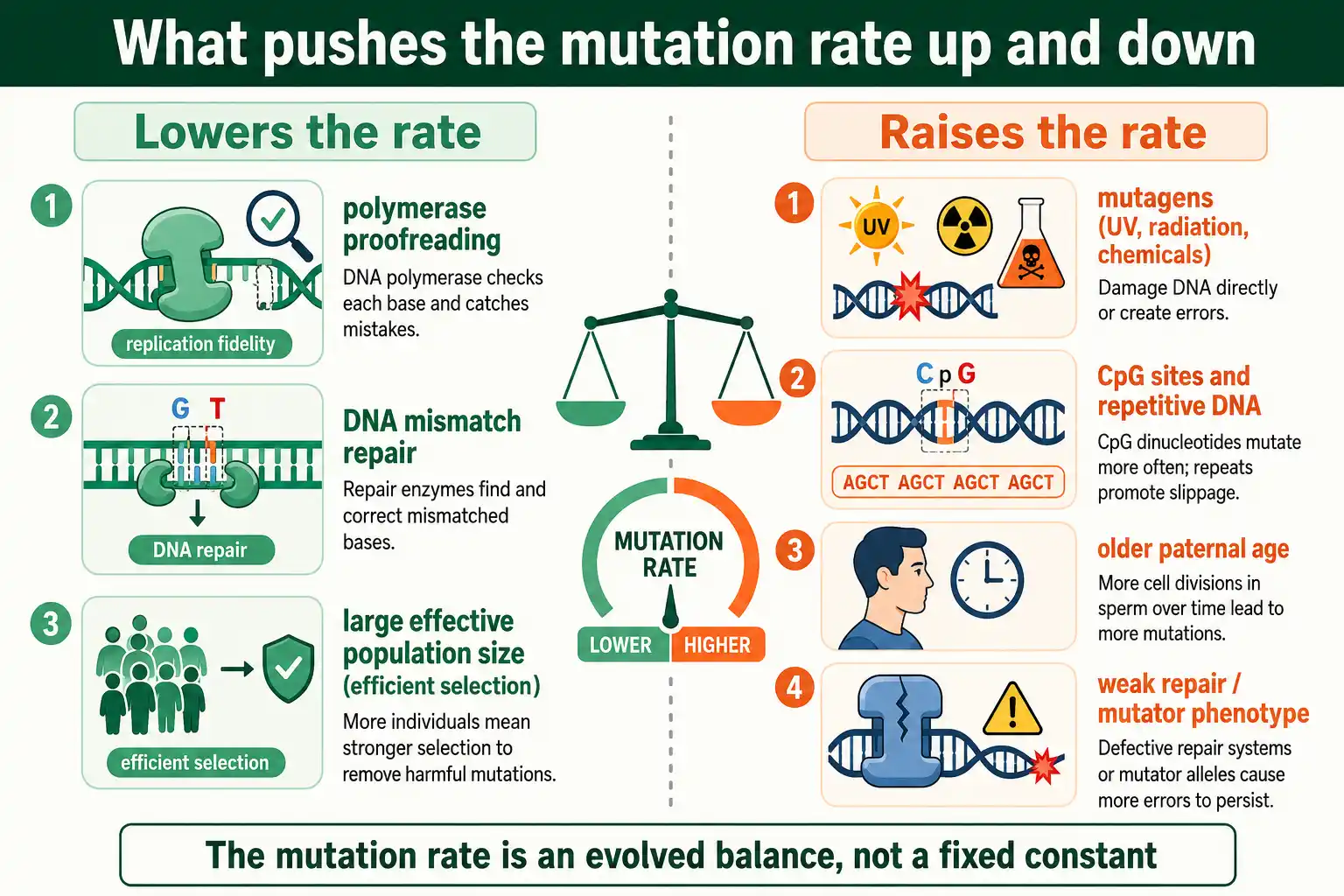
Mutator Phenotypes
Sometimes the mutation rate itself mutates. When a cell or lineage loses part of its replication or repair machinery, its whole-genome mutation rate can jump many-fold, a state called a mutator phenotype. These are important in both evolution and disease.
In bacteria, mutator strains arise when mismatch-repair genes break, raising the mutation rate up to a hundredfold or more. Such strains appear surprisingly often in populations under stress, because a higher mutation rate can occasionally speed adaptation by generating useful variants faster, even though most of the extra mutations are harmful. The mutator allele then hitchhikes along with the beneficial mutations it helped create. This is a rare case where a higher mutation rate is temporarily favored.
In humans, mutator phenotypes drive cancer. Many tumors carry defects in repair or proofreading genes, which sends their somatic mutation rate soaring and accelerates the accumulation of the further mutations that fuel tumor growth. Some inherited cancer syndromes work this way, with a faulty repair gene raising the mutation rate across all of a person's cells. Hypermutated tumors, those with the highest mutation loads, often result from combined proofreading and repair defects, and they have become clinically important because their many mutations can make them more visible to the immune system. The mutation rate, in other words, is not just a population-genetic parameter; its breakdown in individual cells is central to cancer.
Sequence Context and Hotspots
The mutation rate is not uniform across the genome. Some sequences mutate far faster than others, creating mutation hotspots, and the local DNA context is a strong predictor of where mutations land.
The clearest example is the CpG site, a cytosine followed by a guanine. In many organisms, cytosines at CpG sites carry a methyl group, and methylated cytosine spontaneously deaminates to thymine far more readily than ordinary cytosine. This makes CpG sites mutate roughly ten times faster than other positions, so they are the genome's most prominent hotspots. A related general pattern, seen in nearly every species studied, is a bias toward mutations from G or C bases to A or T bases, which steadily shapes genome base composition.
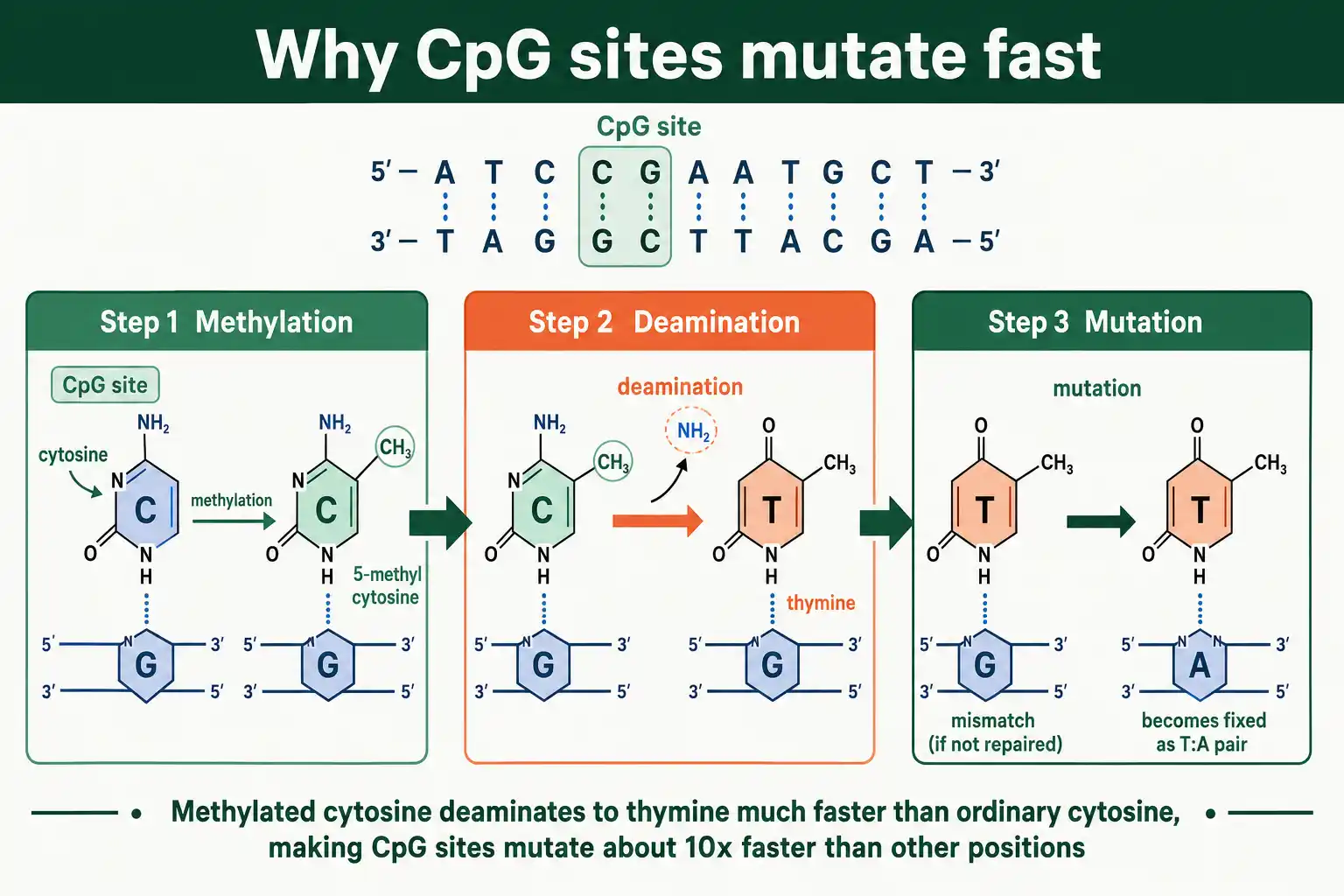
Other context effects exist too. Repetitive sequences, like runs of a single base or short tandem repeats, mutate quickly because the replication machinery slips on them, adding or removing units. Late-replicating regions of the genome tend to have higher mutation rates than early-replicating ones. The upshot is that a single genome-wide average rate hides large local variation, and where a mutation is likely to occur depends heavily on the surrounding sequence.
Parental Age and Sex
In humans and many animals, the mutation rate depends on the age and sex of the parent. This is one of the most striking and well-documented factors, with direct relevance to family planning.
Most human mutations come from the father, and the number rises with his age. The reason is biological: the cells that produce sperm keep dividing throughout a man's life, and every division is a chance for a copying error, so older fathers transmit more mutations. Each additional year of paternal age adds roughly one to two more de novo mutations to the child. Maternal age contributes far fewer new point mutations, because egg cells do not keep dividing the same way, though maternal age raises other risks like chromosomal nondisjunction.
This paternal age effect means the mutation rate is not even a single number within a species. It varies systematically with the father's age at conception, which is part of why the children of older fathers carry a modestly higher risk of certain genetic conditions. The rate quoted for humans, about 1.2 x 10^-8 per base, is an average across typical parental ages, not a universal constant.
The Drift-Barrier Hypothesis
Why does the mutation rate sit where it does, rather than being driven all the way to zero? Selection favors accuracy, since most mutations are harmful, but it cannot perfect it. The leading explanation is the drift-barrier hypothesis.
The logic is subtle. Natural selection does favor lower mutation rates, because fewer harmful mutations means higher fitness, so selection keeps pushing replication and repair toward greater accuracy. But each further improvement yields a smaller and smaller fitness gain. Eventually the advantage of a tiny additional improvement becomes so small that random genetic drift overwhelms it, and selection can no longer push the rate lower. That point is the drift barrier, and it sets the floor on how low the mutation rate can evolve.
The powerful prediction is that the barrier depends on effective population size. In large populations, selection is efficient and drift is weak, so the rate can be pushed very low before hitting the barrier. In small populations, drift is strong and overwhelms selection sooner, leaving a higher mutation rate. Michael Lynch and colleagues developed this hypothesis and showed, in a 2012 PNAS paper on the drift-barrier hypothesis, that across the tree of life species with larger effective population sizes tend to have lower mutation rates, exactly as predicted. This ties the mutation rate directly to population size and drift, the subject of our guides on genetic drift and effective population size.
The hypothesis also explains an apparent paradox. Multicellular eukaryotes like humans, with relatively small effective population sizes, have higher per-base mutation rates than microbes with enormous population sizes, even though the microbes replicate far more often. Under older ideas that mutation rates are set purely by the energetic cost of fidelity, this is hard to explain. Under the drift barrier, it falls out naturally: the microbes' vast populations let selection refine replication accuracy to a degree that drift prevents in smaller eukaryotic populations. The mutation rate, on this view, is less a chemical limit than an evolutionary equilibrium between selection for accuracy and the drift that caps it.
The Factors at a Glance
The factors differ in whether they act on the molecular machinery, the environment, or evolutionary forces. The table organizes them.
| Factor | How it changes the rate | Direction |
|---|---|---|
| Replication fidelity | Polymerase accuracy and proofreading | Better fidelity lowers the rate |
| DNA repair | Mismatch and damage-repair pathways | Better repair lowers the rate |
| Mutagens | UV, radiation, chemicals, reactive oxygen | More exposure raises the rate |
| CpG sites | Methylated cytosine deaminates to thymine | Raises the rate at these sites about tenfold |
| Repetitive sequence | Polymerase slippage | Raises the rate locally |
| Paternal age | More germline cell divisions with age | Older fathers raise the rate |
| Effective population size | Drift barrier on selection for fidelity | Larger populations lower the rate |
The factors fall into three groups. Molecular factors, fidelity and repair, set the baseline machinery. Environmental and contextual factors, mutagens and sequence hotspots, modulate it locally and over a lifetime. And the evolutionary factor, the drift barrier set by population size, explains where the whole rate settles over deep time. Together they explain why mutation rates vary across the genome, between individuals, and across species.
Frequently Asked Questions
What increases the mutation rate?
The mutation rate rises with weaker DNA replication proofreading, defective repair systems, exposure to mutagens like UV light, ionizing radiation, and chemical mutagens, mutation-prone sequences such as CpG sites and repetitive DNA, and older paternal age. At the evolutionary scale, smaller effective population sizes allow higher mutation rates because genetic drift weakens selection for replication fidelity.
Why are CpG sites mutation hotspots?
Because the cytosine in a CpG site is often methylated, and methylated cytosine spontaneously converts to thymine through deamination much more readily than unmethylated cytosine. This makes CpG sites mutate roughly ten times faster than other positions in the genome, so they account for a disproportionate share of point mutations, including many disease-causing ones.
Why isn't the mutation rate zero?
Because selection for ever-greater accuracy hits diminishing returns. Each further improvement in replication fidelity yields a smaller fitness gain, and eventually that gain becomes too small for selection to favor against the random force of genetic drift. This drift barrier sets a floor on how low the rate can evolve, and the floor is higher in small populations where drift is stronger.
What Sets the Rate
The mutation rate is the product of several forces. Replication fidelity and DNA repair set the baseline by determining how many errors are made and corrected. Mutagens and sequence context, especially fast-mutating CpG sites, push the rate up locally and with exposure. Parental age, mainly paternal, raises it within a species. And over evolutionary time, the drift-barrier hypothesis explains why the rate settles where it does, with selection for fidelity opposed by genetic drift in a balance set by population size.
The throughline is that mutation rate is an evolved, variable trait, not a fixed property of chemistry. It differs across the genome, between individuals, and across species, for reasons that span molecular biology, the environment, and population genetics. To see how mutation interacts with selection once mutations exist, our guide on mutation-selection balance shows how the rate sets the equilibrium frequency of harmful alleles.